Alinhamento de Sequências

Neste nosso primeiro post queremos falar sobre um dos procedimentos mais importantes para a análise de sequências biológicas: o alinhamento de sequências. Sem dúvidas realizar alinhamento é um dos procedimentos mais essenciais para a bioinformática e para entender diferentes faces da biologia. Se você é um estudante de bioinformática, é importante que você compreenda o funcionamento dessas ferramentas e saiba como usá-las em suas análises!
Hoje devido à complexidade e a possibilidade de alinhar sequências grandes, os alinhamentos são realizados por programas alinhadores de sequência, que comparam duas ou mais sequências e determinam seu grau de semelhança. Eles são usados em diversas áreas da bioinformática, desde a genômica comparativa até a análise de RNA-Seq para diferentes fins. Com o aumento exponencial no número de sequências biológicas disponíveis, a análise de alinhamentos tornou-se imprescindível para a compreensão da biologia molecular.
Afinal porque alinhar sequências?
Alinhar sequências permite identificar semelhanças e diferenças entre elas. Isso é fundamental para a compreensão da biologia molecular, pois nos ajuda a entender a evolução das espécies, a identificar regiões conservadas que podem ter funções importantes, e a identificar mutações e outros eventos genéticos que podem estar associados a doenças ou características biológicas específicas. Além disso, o alinhamento é um pré-requisito para muitos pipelines, como para a montagem de genomas tanto de novo (contra um genoma de referência) quanto ab initio (sem referência), a análise de RNA-Seq, a identificação de variantes genéticas, filogenômica, dentre outras.
O problema computacional
Dadas duas sequências m e n, existem diversas formas de alinhá-las e quanto maiores forem, mais possibilidades surgem. O número de alinhamentos possíveis para elas é calculado como (m+n)!/m!*n!
Isto significa que para duas sequências de tamanho 11, há 705.432 alinhamentos possíveis, enquanto que para duas sequências de tamanho 100 (ainda muito menor do que muitos alinhamentos que fazemos) as possibilidades crescem para mais que 10^60.
Então como fazemos alinhamentos?
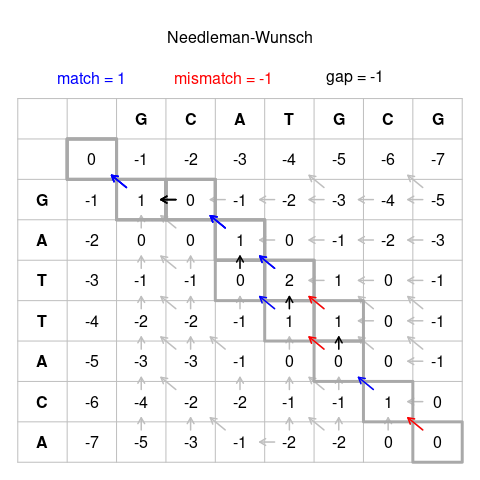
Dois pesquisadores, Saul B. Needleman e Christian D. Wunsch criaram um algoritmo em 1970 que permitia alinhar duas sequências de uma forma computacionalmente possível. A lógica do algoritmo Needleman-Wunsch é utilizada até hoje com algumas adaptações.
De forma breve, o algoritmo Needleman-Wunsch começa por criar uma matriz de pontuação que é preenchida com as pontuações de correspondência, penalidades de gaps e outras penalidades de substituição (mismatch entre nucleotídeos). Em seguida, o algoritmo usa esta matriz para calcular uma pontuação de alinhamento para cada posição possível no alinhamento, utilizando uma fórmula que considera as pontuações de correspondência e as penalidades de lacunas.
Em seguida, o algoritmo realiza um rastreamento na matriz de pontuação para encontrar o caminho com a maior pontuação, o que resulta no alinhamento ótimo global entre as duas sequências.
Esquema do algoritmo Needleman-Wunsch
O algoritmo Needleman-Wunsch ainda é computacionalmente intensivo para sequências muito grandes. Por isso, foram desenvolvidos algoritmos mais eficientes para alinhamento local, como o algoritmo Smith-Waterman.
Hoje, existem diferentes parâmetros que mudam a fórmula das penalidades, levando em consideração diferentes premissas das sequências a serem alinhadas: alinhamentos progressivos, iterativos, local ou global, matriz de substituição de nucleotídeos ou aminoácidos, etc.
Alinhamento em diferentes áreas
Sequenciamento
O alinhamento é uma etapa fundamental em muitos processos de sequenciamento, como o sequenciamento de nova geração (NGS). No sequenciamento NGS, os reads podem ser alinhados contra um genoma de referência para identificar as regiões correspondentes e, assim, ajudar a montar o novo genoma.
Existem diferentes abordagens para o alinhamento de sequências de NGS, mas a maioria dos algoritmos utiliza uma abordagem de alinhamento local. O alinhamento de sequências de NGS é uma etapa crítica em muitos processos de análise de dados, incluindo a detecção de variantes genéticas, a análise de expressão gênica e a identificação de isoformas de mRNA.
Genômica comparativa
Para a genômica comparativa, a primeira etapa consiste em alinhamentos das proteínas de cada espécie e, de forma bem simplificada, a separação das mais similares em famílias gênicas para posterior análise. As famílias gênicas também precisam ser alinhadas para quaisquer análises filogenéticas posteriores.
Metagenômica
De forma semelhante ao sequenciamento,quando se trabalha com metagenômica um dos passos necessários é alinhamento dos reads contra bancos de dados de referência para identificar as espécies presentes na amostra.
Programas de alinhamento
Existem inúmeros programas de alinhamento disponíveis, mas alguns são mais comuns e estabelecidos do que outros. Alguns dos programas mais utilizados são:
Conclusões
O alinhamento de sequências é uma etapa crítica em muitos processos de bioinformática e a otimização algorítmica permitiu diversos avanços na bioinformática. A escolha do algoritmo de alinhamento e dos parâmetros adequados para a análise pode ter também um grande impacto nos resultados finais e deve ser considerada durante as análises.